
相关热词搜索: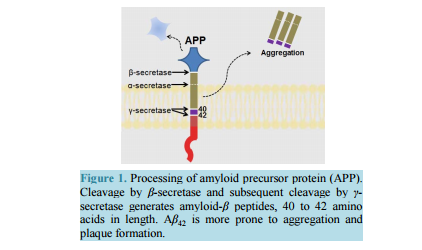 Strategizing the Development of Alzheimer’s Therapeutics Justin Davis, Robin Couch Department of Chemistry and Biochemistry, George Mason University, Manassas, VA, USA Email: [email protected] Received 10 June 2014; revised 25 July 2014; accepted 5 August 2014 Copyright © 2014 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Alzheimer’s Disease is a complex, progressive condition with symptoms that do not reveal themselves until significant changes to neuronal morphology have already occurred. The delayed manifestation of cognitive decline makes determination of the true etiological origins difficult. As a result, identification of ideal drug targets becomes seemingly impossible. The existing treatments for Alzheimer’s Disease may temporarily suppress the rate of cognitive decline, but do little to slow or halt neuronal decay. While many believe that the current approaches to identifying a cure for the disease are too narrow-minded, focusing heavily on the physical manifestations of the diseased brain such as amyloid plaques and neurofibrillary tangles, this review asserts the status of Alzheimer’s research as rational and multi-faceted. Keywords Drug Discovery, Alzheimer’s Disease, Target Identification, Therapeutics, Review 1. Introduction Alzheimer’s Disease (AD), the most common form of dementia, involves the progressive deterioration of cognitive and functional abilities, ultimately leading to the loss of bodily function and death. According to the World Health Organization, dementia affects 35.6 million people worldwide, with the figure expected to double by 2030 and triple by 2050 in the absence of a new therapy capable of preventing, halting, or slowing disease progression. Alzheimer’s Disease accounts for up to 70% of these cases, with an annual estimated worldwide cost of $600 US billion [1] [2]. In the United States, one in nine people over the age of 65 is believed to suffer from AD [3]. The earliest symptoms of AD include deficits in working memory and concentration. Visuospatial and visuoconstructional deficiencies as well as naming and language impairments also appear early in the course of the J. Davis, R. Couch 108 disease [4]. As AD progresses, changes in social and emotional behaviors occur, including agitation, psychosis, and depression. Later stages of AD are marked by intermediate and long-term memory loss, loss of self-sustainnability, and changes in sleep cycle. The final stages of AD entail the loss of control over bodily function and may often be characterized by a vegetative state [4]. Upon onset, life expectancy is reduced to a range of 4 to 12 years [4], correlating to a 67% reduction in expected life span for diagnoses at age 65 [5]. Alzheimer’s Disease is the sixth leading cause of death in the United States, causing over 80,000 deaths in 2010 [3]. The pathogenesis of Alzheimer’s Disease is still being elucidated and is often characterized by the development of beta-amyloid aggregates, tau hyperphosphorylation, changes in neuronal glucose metabolism, and neurodegeneration. The breakdown of neural connections eventually leads to dementia and loss of cognitive function. Therapeutics currently approved for the treatment of Alzheimer’s Disease help to temporarily mitigate symptoms and improve quality of life by affecting neurotransmission; however, these drugs are not disease-modifying and fail to address the etiological underpinnings of the disease. The following review will detail the current treatments for Alzheimer’s Disease and will present strategies for developing new, disease-modifying treatments based on various elements of known AD pathology. 2. Current Therapeutics: Pharmaceutical Treatment of Neurotransmitter Signaling Currently, the four FDA approved medications for AD treat the symptoms of the disease, mainly by acting on the neurotransmitter signaling. The hallmarking memory deficit of AD was linked to a substantial loss of cholinergic neurons in the basal forebrain and cerebral cortex as early as the 1980s [6] [7]. The noted deficit in cholinergic synapses led to the development of therapeutic agents designed to restore cholinergic neurotransmission via inhibition of acetylcholinesterase (AChE), an enzyme that hydrolyzes free acetylcholine in the synapse. While these agents have demonstrated the ability to slow the rate of cognitive decline, they have little effect on the pathogenesis of AD. Galantamine, a phenanthrene, is the weakest of the approved acetylcholinesterase inhibitors (IC50 400 - 800 nM [8] [9]) and is a competitive, reversible inhibitor of the enzyme [10]. The compound has also been shown to allosterically agonize the nicotinic acetylcholine receptor at low concentrations, with antagonistic effects at higher concentrations, likely through a competitive mechanism [11] [12]. This dual mode of enhancing acetylcholine signaling likely offsets its lower binding affinity for AChE, as galantamine produces similar positive effects on cognition, behavior, and general function when compared to the other approved acetylcholinesterase inhibitors [13] [14]. Donepezil, a piperidine, is a highly specific, non-competitive, reversible AChE inhibitor (IC50 6.7 nM [15]) that fits within the active site gorge of AChE without directly interacting with the catalytic triad of the enzyme [16]. The structural differences between donepezil and the other inhibitors contributes to its longer half-life (-73 hours versus 4 - 6 for rivastigmine and galantamine) [14]. In comparison with placebo, donepezil demonstrates a statistically significant improvement in cognitive function as well as positive behavioral modification [14]. Rivastigmine, a carbamate derivative, is a non-competitive, reversible AChE inhibitor (IC50 4.4 nM [15]) that inhibits both acetylcholinesterase and butyrylcholinesterase with similar affinity [17]. Rivastigmine remains bound to the enzyme for a greater amount of time than the other AChE inhibitors (10 hours versus minutes for donepezil) [18], as its carbamate moiety remains bound in the esteratic site of the enzyme following hydrolysis [18]. Rivastigmine generates improvements in cognitive function at low doses of 1 - 4 mg/day and highdoses of 6 - 12 mg/day. However, the higher doses required to produce enhancements in global assessments also correlate with a higher rate in discontinuation due to adverse effect [14]. Most comparative analyses investigating the effectiveness of the three acetylcholinesterase inhibitors suggest that all three compounds have similar efficacy in regards to cognitive function, behavior, and global outcome [14]. Meta-analysis performed by Hansen et al. in 2008 [19] proposes a greater behavioral response for patients treated with donepezil and a better global assessment with donepezil or rivastigmine, although the differences are minute. Overall, the three acetylcholinesterase inhibitors temporarily stabilize the rate of behavioral, cognitive, and functional decline. These effects last for a range of six months to one year, before cognition falls below baseline levels [20]. The remaining FDA approved treatment for Alzheimer’s disease, memantine, affects glutamate signaling through uncompetitive antagonism of NMDA (N-methyl-D-aspartate) receptors (IC50 0.3 - 10 μM, depending on J. Davis, R. Couch 109 membrane potential [21] [22]). The open channel blocker has a limiting effect on Ca2+ flux, with potential therapeutic effects of suppressing long term potentiation and limiting excitotoxicity [21]. Memantine also has a similar inhibitory effect on both α7 and α4β2 nicotinic acetylcholine receptors (IC50 - 10 μM [21] [22]), with opposite effects on cholinergic transmission than the acetylcholinesterase inhibitors approved as AD therapeutics. The compound shows efficacy in patients with moderate to severe AD when used as a monotherapy [23] [24] or in conjunction with the approved acetylcholinesterase inhibitors [24]. At least 24 compounds targeting acetylcholine signaling and 9 compounds targeting glutamate signaling (through NMDA or AMPA receptors) had been evaluated in clinical trials for Alzheimer’s as of May 2014. Additionally, at least 29 dopamine or serotonin modulators have been evaluated as well. In total, approximately one third of all clinical trials (phase I-IV) for the treatment of Alzheimer’s Disease target neurotransmitter signaling in the brain. These statistics suggest that while Alzheimer’s is often described by the etiological issues of amyloid plaques and tau hyperphosphorylation, the treatment of its symptomatic deficits in memory and cognitive decline is still a key component of attacking the disease and its symptoms. 3. Strategizing Therapeutics: The Amyloid Hypothesis 3.1. General Background Alzheimer’s Disease, as it was first described by Dr. Alois Alzheimer in 1907, is most often characterized by the accumulation of beta-amyloid plaques derived from the aggregation of small amyloid-β (Aβ) peptides in the brain. The amyloid-β peptide is generated by cleavage of membrane-bound amyloid precursor protein (APP), a protein involved in synaptogenesis [25]. The Aβ peptide is excised from APP holoprotein by β-APP cleaving enzyme (BACE or β-secretase) and γ-secretase [26] [27]. β-secretase acts extracellularly on APP, releasing a large fragment (sAPPβ) to the extracellular environment. The remaining 99 amino acid fragment (CTFβ) remains temporarily bound to the membrane [27]-[29]. The subsequent intracellular action of γ-secretase on CTFβ results in the generation of fragments Aβ40 and Aβ42, depending upon the site of cleavage [27]-[30] (Figure 1). The conformation of the longer fragment, Aβ42, differs from that of Aβ40 [31]-[33], causing it to be the “stickier” of the two aggregation-prone beta-amyloid peptides. As a result, Aβ42 is more often implicated in the formation of the amyloid plaques [30]. In healthy inpiduals, γ-secretase generates Aβ40 8 to 9 times more often than Aβ42 [27]-[34]. Additionally, neither β-amyloid is the most prevalent APP cleavage product found in the brain due to APP processing by α-secretase [27] [34] [35]. Although β-secretase activity and its downstream products are significant due to their roles in amyloid plaque formation, β-secretase acts on only 10% of the total APP in healthy cells [27] [35]. Most often, APP is cleaved by α-secretase, generating sAPPα extracellularly and an 83 amino acid fragment, CTFα [26] [27] [35]. Intracellular cleavage of CTFα by γ-secretase generates a p3 fragment that is not prone to plaque formation in the fashion of Aβ [27]. However, while the amount of Aβ42 present in healthy brains is minimal, AD is marked by a suppression of α-secretase activity and a gain in β-secretase activity that enhances plaque formation through increased production of amyloid-β [35]. The driver for this change in relative secretase activities is unknown. Figure 1. Processing of amyloid precursor protein (APP). Cleavage by β-secretase and subsequent cleavage by γ- secretase generates amyloid-β peptides, 40 to 42 amino acids in length. Aβ42 is more prone to aggregation and plaque formation. J. Davis, R. Couch 110 Aggregation of Aβ occurs in two phases, with soluble multimers preceding the development of fibrillar deposits [36] [37]. The two multimeric forms trigger different toxic responses and are recognized by different antibodies [27]. The exact mechanism of action for Aβ induction of neurodegeneration is unknown, but smaller, soluble Aβ aggregates are believed to be the form most relevant to amyloid related toxicity in AD [27] [37]. Additionally, Aβ aggregates can deposit in the walls of blood vessels, causing intracerebral hemorrhaging [38]. Most believe that the development of beta-amyloid aggregates is the initiating step in the disease, and thus, tau hyperphosphorylation, excitotoxicity, changes in glucose metabolism, and neurodegeneration may all be downstream results of amyloid-β accumulation. Prevention of amyloid-β accumulation requires the degradation or clearance of the peptide. The enzymes neprilysin (NEP) and insulin degrading enzyme (IDE) have been implicated as the proteins responsible for degrading Aβ in the cell [27] [39] [40]. Neither protease is specific for Aβ, and IDE shows 20-fold greater binding affinity for insulin than for Aβ [27] (thus, insulin may act as a competitive inhibitor of IDE degradation of Aβ, serving as a potential link between insulin-related diseases and AD). Much Aβ remains undegraded, and some is transported through the blood brain barrier by low-density lipoprotein receptor-related protein (LRP) on the brain side and the receptor for advanced glycation end products (RAGE) on the blood side [27] [41]. Aβ that remains unaffected by degradation and transport has greater chance to form aggregates. The perceived significance of amyloid-β to AD was heightened by the discovery of genetic mutations and risk alleles that relate directly to amyloid-β and its clearance from the cell. Autosomal dominant mutations implicated in familial, early onset AD, have been detected in genes for presenilin 1 and 2 (PSEN 1 and PSEN 2) [26] [42] [43], which code for the catalytic unit of the γ-secretase complex. The mutations of PSEN 1 and PSEN 2 alter the cleavage activities of γ-secretase, resulting in a higher ratio of Aβ42 to Aβ42 [26] [42]. Autosomal dominant mutations in the gene for amyloid precursor protein that cause changes in processing have also been implicated in promoting the generation of Aβ42 [44] [45]. Additionally, three different apolipoprotein E (APOE) alleles are common in the population. APOE2, APOE3, and APOE4 have been correlated to AD with APOE2 delaying the onset of disease, APOE3 being neutral, and APOE4 decreasing the age of onset [26]. The mechanism of action is believed to involve direct removal of amyloid-β [46], as APOE2, APOE3, and APOE4 have decreasing effectiveness in Aβ clearance [26] [47]. As the only known genetic mutations and risk alleles for AD relate directly to amyloid-β, and not to other known features of AD, inhibiting Aβ production and preventing its aggregation is a rational approach to treating the disease. 3.2. Directed Therapeutics: Gamma-Secretase Inhibition and Modulation Gamma secretase has been evaluated as a target to minimize beta-amyloid production with minimal success. The aspartylintramembranal protease is composed of Aph1, nicastrin, Pen2, and presenilin, the gene product implicated in two of the three major genes associated with familial AD [48]. Presenilin serves as the catalytic component of γ-secretase. However, the protease also acts on other transmembrane proteins, including p75 neurotrophin receptor, ErbB4, N-cadherin, and Notch [49] [50]. Proteolytic cleavage of Notch by presenilin promotes its nuclear translocation, initiation of signal transduction cascades, and transcription regulation, inhibiting downstream modulation of cell differentiation and proliferation [51] [52]. As expected, γ-secretase inhibitors (GSIs), such as MK-0752, have been evaluated in clinical trials as anti-cancer agents due to its depressive effects on Notch [53]. Complete inhibition of γ-secretase functioning, through binding to presenilin fragments involved in protease activity, results in dramatic side effects. Semegestat, the poster-drug for detractors of the amyloid hypothesis, advanced to phase III clinical trials as a γ-secretase inhibitor, but led to significant worsening of cognitive function despite reducing beta-amyloid load [50]. The decline in cognitive function may be linked to observed changes in hippocampal signaling [54]. The mechanism is unknown, but may be attributed to downstream effects on presenilin’s other protease targets, including Notch, which has been implicated in the regulation of dendritic development [55] and axon arborization (branching) [56]. Noted hematologic toxicity, skin toxicity, and gastrointestinal toxicity, likely induced by changes in intestinal epithelium due to Notch inhibition [57], brought a premature end to the clinical trial, and a similar fate was shared by avagacestat, another γ-secretase inhibitor. Avagacestat was also linked to an increase in nonmelanoma skin cancer during phase II clinical trials [58], causing further debate as to the viability of γ-secretase as a suitable drug target for AD. These results have J. Davis, R. Couch 111 brought about a change in the design of γ-secretase inhibitors, from compounds aimed at total complex inhibition, to compounds designed to modulate γ-secretase. These γ-secretase modulators (GSMs) inhibit γ-secretase through allosteric mechanisms that permit the other actions of presenilin. The overarching aim of GSMs is to alter the γ-secretase complex in a way that favors the production of shorter forms of beta-amyloid that are less prone to aggregation, such as Aβ38,without interfering with γ-secretase action on other targets such as Notch. In one potential mechanism, noncompetitive binding to a novel site on γ-secretase induces a conformational change that shifts the cleavage site [59]. Alternatively, the compound may act directly with APP, altering the availability of the Aβ42 and Aβ40 cleavage sites [48] [60]. Over the past decade, several nonsteroidal anti-inflammatory drugs (NSAIDs), including ibuprofen, have been evaluated as a treatment for AD due to an observed reduction in AD prevalence amongst NSAID users. These compounds, serving as the first generation of GSMs, demonstrated effects of γ-secretase modulation in vitro, with a decrease in Aβ42 correlating to an increase in Aβ38. Tarenflurbil, R-fluriprofen, failed phase III clinical trials in 2009, showing no effect on cognitive decline [61]. Although the compound showed an effect on Aβ42 in cell based assays, similar effects were not observed in patients despite dose dependent blood-brain barrier penetration [62]. More recent methods of designing GSMs have involved modifications to existing NSAID structure as well as designing new compounds altogether, with the ultimate goal of reducing Aβ42 levels in the human brain. In general, γ-secretase modulators have reduced potency compared to γ-secretase inhibitors in terms of overall reduction of Aβ42 [63]. As such, the design of blood brain barrier-permeable molecules that exhibit minimal toxicity at concentrations sufficient for preventing Aβ42 production is the greatest barrier in the γ-secretase directed therapies. 3.3. Directed Therapeutics: Beta-Secretase Inhibitors Another avenue of reducing amyloid load in the brain is by limiting Aβ production using β-secretase (BACE-1) inhibitors. These compounds reduce the amount of Aβ detected in plasma and cerebrospinal fluid (CSF) after administration [50] [64] [65] and have generated cognitive improvements in mouse models [66]. The side effects of BACE-1 inhibition have been debated, with abnormalities in BACE-1 negative mice ranging from minimal [64] [67] to the development of memory deficits, seizures, hypomyelination, and changes in axonal guidance [68]-[70]. BACE-1 inhibitors have become prevalent in clinical trials since the first phase I study of CTS21166 in 2008. This compound caused a dose-dependent depletion of plasma Aβ by up to 80%, with full recovery occurring 144 hours after administration [64]. A second BACE-1 inhibitor, LY2886721, demonstrated reductions in Aβ40 and Aβ42 in a phase I study; however, phase II trials were terminated prematurely due to toxicity in the liver. A relationship between the BACE-1 inhibition mechanism and liver-related side effects is yet to be identified [65]. MK-8931, the first BACE-1 inhibitor to enter a phase III trial, should demonstrate the effects of BACE-1 inhibitors on cognitive function. The compound has, like the other clinically tested BACE-1 inhibitors, led to reductions in Aβ following administration [50]. MK-8931 is being evaluated in cohorts with mild and prodromal AD, and the results should shed light on the potential advantages of targeting Aβ at early disease stages. In the past five years, at least four BACE-1 inhibitors have completed clinical trials as Alzheimer’s therapeutics and two are currently in testing. 3.4. Directed Therapeutics: Active Immunization Active immunization against amyloid-β has been attempted since the late 1990s with limited success. Theoretically, the strategy would allow a patient’s immune response to produce oligoclonal antibodies for beta-amyloid that are persistent. The first major attempt, AN1792, utilized the complete Aβ42 peptide in combination with the QS-21 adjuvant to elicit an immune response capable of clearing amyloid-β. In the case of active and passive immunization, Aβ clearance appears to depend on a microglial FcR dependent mechanism that stimulates phagocytosis [71]. Mouse models showed not only a reduction in amyloid load, but also a suppressive effect on cognitive decline, corresponding to a reduction in neuritic dystrophy and angliosis [72]. However, Phase II clinical trials in 2001 were unsuccessful. Although the treatment successfully caused a decrease in total amyloid-β load (analyzed post-mortem), the active immunization method yielded no effect on cognitive performance or survival over the 6 year follow-up period. Additionally, meningoencephalitis occurred in 18 of the 80 patients studied, causing the trial to be ceased prematurely [50] [73]. More recent approaches to active vaccination have utilized only the N-terminal fragment of amyloid-β and have varied the use and type of adjuvants in order to promote amyloid clearance without inducing excessive in- J. Davis, R. Couch 112 flammation. UB 311 employs two synthetic peptides linked to the N-terminal 14 amino acid fragment of amyloid-β and is provided with a mineral salt adjuvant. Mouse studies showed diminished amyloid-β load in the cortex and hippocampus and corresponding improvements in learning and short term memory. Initial results of a clinical trial involving 19 patients revealed elevated antibody titers over a 48 week period, as well as measurable improvement in cognitive function, with no reported brain inflammation noted thus far [74]. A second vaccine, AC-24, utilizes an N-terminal 15 amino acid fragment in a liposomal surface. AFFiris has employed an approach of using short peptide fragments of 8 amino acids or less in order to minimize T cell response. CAD106 also incorporates this tactic and successfully causes an increase in antibody titers without eliciting T cell response [75]. The vaccination approach for reducing amyloid load is becoming more prevalent, with the focus resting on maximizing antibody generation and amyloid clearance whilst minimizing inflammation. Adjuvant selection, fragment choice, and the stage of AD in which vaccination is delivered will all play roles in the success of this treatment method. 3.5. Directed Therapeutics: Passive Immunization Passive immunization using humanized IgG antibodies against beta-amyloid provides a second option to promote Aβ clearance without eliciting T-cell mediated inflammation. The most glaring challenge in a passive antibody mediated therapy would likely by delivering the antibody to the brain across the blood brain barrier. As early as 2000, passive immunization was displaying efficacy in mouse models by reducing amyloid load, limiting neuritic dystrophy, and producing a marked improvement in cognitive function [76]-[78]. Successes in the mice model go against expectations for blood brain barrier permeability. The first attempts at passive immunization resulted in elevated levels of plasma Aβ and little Aβ bound to antibody in the brain. The phenomenon lead to the development of the “peripheral sink” hypothesis. In this mechanism, equilibrium draws the amyloid-β peptide into the plasma after plasma-Aβ levels are reduced by the antibody [76]. This result was observed not only in the mouse model, but also in the case of bapineuzumab, the first monoclonal anti-Aβ antibody to reach Phase II clinical trials. Bapineuzumab, a humanized monoclonal anti-Aβ antibody directed at the N-terminus of Aβ, exhibits ApoE dependent reduction in beta-amyloid, with carriers of ApoE4 showing minimal response [50] [76]. Several cases of mild vasogenic edema were observed, more commonly in ApoE4 carriers, but these cases were not severe enough to cause trial cessation. The correlation between the peripheral sink hypothesis and vasogenic edema do suggest the possibility of an inflammation-based mechanism of action that depends upon enhanced blood brain barrier permeability. In terms of cognition, a minimal clinical effect was observed, with a 5% - 6% improvement compared to placebo at the end of 78 weeks (ADAS-Cog and DAD scores respectively) [76]. Phase III clinical trials of bapineuzumab have been discontinued due to failure to show significant changes in cognitive end points, but the compound did elicit a decrease in amyloid burden and CSF phosphorylated tau [79]. The other passive immunotherapy to have completed phase III clinical trials, Solanezumab, is directed at a central epitope of Aβ, favoring the removal of free Aβ or small soluble fibrils over plaques. This compound produced no effect on phosphorylated tau and failed phase III due to inability to meet cognitive end points. More recent attempts at passive immunization are derived from monoclonal antibodies developed against protofibrillar species (BAN2401, Crenezumab, SAR228810) [79]. Clinical outcome data from trials involving these treatments should be released in the upcoming years, providing more evidence as to the potential efficacy of passive immunization. 3.6. Directed Therapeutics for Beta-Amyloid: Conclusion Thus far, beta-amyloid directed therapies have failed to slow or halt the progression of Alzheimer’s Disease. However, the recent progress in immunization and secretase-inhibition do provide hope for a potential beta-amyloid targeted cure. Development in active immunization, passive immunization, γ-secretase inhibition, and β-secretase inhibition is currently ongoing, providing a multifaceted approach for preventing beta-amyloid accumulation. 4. Strategizing Therapeutics: Tau Hypothesis 4.1. General Background Failures of amyloid-based treatments to have a significant impact on the progression of Alzheimer’s Disease J. Davis, R. Couch 113 have called into question the role of amyloid plaque formation as a causative agent of AD. Clinically, loss of brain mass and the progression of dementia correlate only weakly to the quantity and distribution of amyloid-β in the brain, and amyloid load may be present in up to 30% of the elder population with no signs of the Alzheimer’s phenotype [27] [80] [81]. Instead of being the driving force behind the AD neurodegeneration, many have postulated that the formation of amyloid plaques is just one of many secondary events that occur in the progression of the disease. The Tau hypothesis seeks to provide an alternative rationale, attributing Alzheimer’s Disease to the formation of neurofibrillary tangles composed of hyperphosphorylated tau [26] [27] [30]. The causes of excessive tau phosphorylation are still unverified, but toxicity induced by Aβ, oxidative stress, and inflammation may induce upregulation of tau kinases, downregulation of phosphatases, and modification to tau directly (post-translational modifications of tau are discussed in the next section) [82] [83]. Tau is a soluble microtubule-associated protein and has stabilizing effects on microtubulin assembly [84]-[86]. Hyperphosphorylation of tau, an indicator of AD, inhibits the association of tau with the microtubules and induces the formation of tau aggregates [87]-[89]. The microtubule network plays a critical role in neuronal morphology and the transport of signaling molecules, trophic factors, and organelles [85] [86]. Tau mutations and hyperphosphorylation have been correlated to a reduction in microtubule density and fast axonal transport in mouse models [83]. In a recent study, providing a microtubule-stabilizing drug to tau-transgenic mice with microtubule and motor deficits led to improvement in microtubule density and fast axonal transport as well as an improvement in motor function [83]. The gene for tau (tubulin-associated unit) is composed of 16 exons. The protein is generally described as four segments (Figure 2): an acidic N-terminal segment produced from exons 1 - 5, a proline rich region from exon 7 and part of exon 9, the microtubule association region composed of exons 9 - 12, and a C-terminal portion from exon 13. Products from exon 6, 8 and 14 are not observed. Exons 2, 3, and 10 undergo alternative splicing, resulting in six observed isoforms with varying affinity for microtubulin. These isoforms are designated 4R/2N, 4R/1N, 3R/2N, 4R/0N, 3R/1N, and 3R/0N. R describes the exons in the repeat domain region, with 4R designating the presence of exon 10 and 3R designating absence. N describes the Nterminal region, with 2N containing both exon 2 and 3, 1N containing only exon 2, and 0N containing neither exon 2 nor exon 3 [82] [90]-[92]. The repeat domains, responsible for microtubule binding, contain the conserved sequence motif, KXGS. The serine residue in this motif is prone to phosphorylation, resulting in destabilization of the tau protein. Tau expression appears to be dynamic, with 3R tau being more prevalent during development, allowing for enhanced plasticity corresponding to decreased cytoskeleton support. Adult neurons coincide with a closer to 1:1 ratio of 4R:3R and decreased hyperphosphorylation, providing more tau-assisted cytoskeletal stabilization. In disease, a change in 4R:3R ratio is observed [92] [93]. The effect of tau hyperphosphorylation on the microtubule network is one of only two potential causes of tau induced neurodegeneration. Neurofibrillary tangles occur when hyperphosphorylated tau forms aggregates with tau, microtubule associated proteins 1 and 2, and ubiquitin [82] [88]. Straight or paired helical forms of these aggregates exist with a notable toxic effect on the cellular environment [82]. The abnormally phosphorylated tau, PHFtau, can aggregate to form paired helical filaments [94] [95]. However, only 10% of PHFtau is found in neurofibrillary tangles, with the remaining portion deposited in neuronal processes, appearing as dystrophic neuritis with swollen axons and dendrites [94]. Recent animal model studies suggest that microtubule stability is sufficient for cognitive improvement, regardless of the continued presence of neurofibrillary tangles [94]. This evidence points to microtubule dissociation as the key component of disease progression resulting from hyperphosphorlyated tau. 4.2. The Role of Kinases and Phosphatases in AD Pathology Three classes of tau kinases phosphorylate the tau protein, including tyrosine-specific kinases, proline directed kinases (PDKs), and non-proline directed kinases. The major proline directed kinases acting on tau include glycogen synthase kinase (GSK-3β), cyclin dependent kinase (CDK5), and mitogen-activated protein kinases (MAPKs). Relevant non-PDK kinases are tau-tubulin kinase, casein kinase, dual-specificity tyrosine phosphorylation and regulated kinase (DYRK1A), microtubule affinity-regulating kinases, PKAA, PKB/Akt, PKC, PKN, and Calmodulin-dependent protein kinase (CaMKII) [92]. Hyperphosphorylated tau contains up to 10 phosphates per molecule, representing a 3 to 4 fold increase compared to tau in healthy inpiduals. Of the 85 serine, J. Davis, R. Couch 114 Figure 2. The largest tau gene product, 4R/2N, is indicated by the presence of exon 10 and exons 2 and 3. Six isoforms of the gene exist. The 4R/1N isoform is marked by the absence of exon 3 and 4R/0N contains neither exon 3 nor exon 2. The 3R isoforms have lower affinity for microtubulin and are marked by the absence of exon 10. threonine, and tyrosine residues that could serve as targets for phosphorylation by esterification, as many as 45 are exclusive targets for hyperphosphorylation in AD [96]-[100]. GSK-3β, CDK5, CK1, and PKA demonstrate the ability to generate all but eleven of these phosphorylation events, with much overlap in coverage amongst the kinases [96] [99] [101]-[103]. In AD, changes in expression and activity of GSK-3β, CDK5, DYRK1A, p38, and CK1 have been reported [92]. The sequence of phosphorylation events could provide insight into potential therapies. Phosphorylation by DYRK1A and CDK5 precede subsequent GSK-3β phosphorylation in vitro. Additionally, CK1 and c-Abl activate CDK5 phosphorylation events [93]. PKB, PKC, and PKN appear to inhibit phosphorylation by GSK-3β [104]. GSK-3β receives enhanced attention because of its implication in long term potentiation [100] [105] [106]. The kinase plays a role in NMDA receptor trafficking and therefore glutamate signaling, implying a potential mechanism for excitotoxicity [100] [107]. Additionally, a region of presenilin 1 that interacts with GSK-3β exhibits binding affinity for the microtubule binding domains of tau, providing a mechanism in which presenilin could facilitate an interaction between tau and the kinase and suggesting another means for presenilin mutations to induce neurodegeneration [90]. GSK-3β may also play an integral role in amyloid-induced tauopathy. In mouse models, amyloid peptides induce tyrosine autophosphorylation of GSK-3β at Y216, activating the kinase [100] [108]. In the frontal cortex of the AD brain, pY216-GSK3 has been observed as co-localized with tau protein, supporting this model of GSK activation leading to tau hyperphosphorylation [109] [110]. Although some debate surrounds whether or not the development of beta-amyloid deposits absolutely occurs before tauopathy in every case of AD [111] [112], models suggest that tau reduction prevents amyloid-induced excitotoxicty [113] [114] and defects in axonal transport [115]. Phosphatases, specifically protein phosphatase-2A (PP2A), play a critical role in tau hyperphosphorylation. The region of tau responsible for binding microtubules shows high affinity for PP2A, alluding to a mechanism in which unbound tau serves as a substrate for PP2A dephosphorylation, promoting subsequent binding to microtubules [90]. The Alzheimer’s brain exhibits a 50% reduction in PP2A activity and a 20% increase in PP2A inhibitors PP2A 1I and PP2A 2I [92] [116]. Expression of mRNA coding for the regulatory subunits and catalytic subunit of PP2A is downregulated in the hippocampus [117]. In model systems, the addition of PP2A reduces tau aggregates and encourages tau binding to microtubules [92] [118]. Methylation of the catalytic subunit of PP2A is required for activity, yet the responsible methyltransferase (LCMT-1) is under expressed in AD brains [119] [120]. Additionally, PP2A phosphatase activator, an enzyme that activates PP2A by removing an inhibitory phosphate, is also downregulated in AD [120] [121]. Glycosylation of tau, particularly O-glycosylation by N-acetyl-glucosamine, is another posttranslational modification that may impact tau aggregation and association with microtubules. Conflicting descriptions of the glycosylation state of tau and its ramifications exist in the literature. Multiple studies have described glycosylation of tau in PHF tangles with no glycan observed in normal tau [122] [123]. These reports also suggest that glycosylation favors association into filaments, and that deglycosylation is sufficient for conversion into linear bundles, with dephosphorylation and deglycosylation having a restorative effect on tau binding to microtubulin [122] [123]. Data also indicates that glycosylation of tau in the AD brain precedes tau hyperphosphorylation [124] [125]. Such a result might imply that glycosylation makes tau a better substrate for kinases or a poorer substrate for phosphatases, and in vitro assays do support this hypothesis [125]. However, recent evidence suggests that O-linked N-acetylglucosamine is more prevalent in healthy controls than in AD brains, and that glycosylation has an inhibitory effect on phosphorylation [126] [127]. In 2012, Yuzwa et al. performed a mouse model study [128] using an inhibitor of O-GlcNAcase that resulted in increased levels of O-glycosylated tau, decreases in tau aggregation, and decreases in neuronal cell loss. Interestingly, no changes in tau phosphoryla- J. Davis, R. Couch 115 tion were observed. The results conflict with evidence suggesting that glycosylation promotes aggregation; however, the authors’ in vitro analysis supports their claim of O-GlcNAc inhibiting oligomerization of tau. While many advocate the possibility of O-GlcNAcase inhibitors as a potential therapeutic for the treatment of AD, questions remain as to the consistency of tau glycosylation effects in AD etiology. 4.3. Directed Therapeutics: Kinase Inhibition The use of GSK-3β inhibitors as a means of treating AD is being evaluated. In mouse models, inhibition of GSK-3β resulted in reduced tau phosphorylation, tauopathy, neurodegeneration, neurofibrillary tangle formation, and neuronal loss [93]. Two specific GSK-3β inhibitors, NP031112 and tideglusib, have completed phase II trials with results yet to be published. In a pilot study, tideglusib appeared to elicit a positive cognitive response with minimal toxicity, although sample size was inefficient to determine true efficacy [129]. Cerebrolysin, a compound with regulatory effects on GSK-3β and CDK5 has also been involved in clinical trials as a combinatorial therapy with the approved acetylcholinesterase, donepezil. In a small sample size, cerebrolysin showed similar improvements as donepezil in cognitive performance, with a combinatorial approach showing enhanced gain over either inpidual treatment [130]. A c-Src/Abl kinase inhibitor, saracatinib, is currently in phase I trial, and more work targeting tau-related kinases is ongoing. 4.4. Directed Therapeutics: Tau Aggregation Inhibition An alternative method of treating tauopathy is through the use of tau aggregation inhibitors. In vitro assays and mouse model studies propose methylthionium chloride (methylene blue) as a compound capable of reducing aggregation of phosphorylated tau [131]-[133]. TauRx has developed multiple aggregation inhibitors that are derivatives of methylthionium chloride. The first of these compounds, TRx0014 has completed phase II clinical trials. Although the results pertaining to tau aggregation have yet to be published, preliminary data suggests a statistically significant delay in the rate of cognitive decline [134]. However, methylene blue has been indicated as an effector of butyrylcholinesterase activity [135], monoamine signaling [136] [137] and glutamate signaling [131] [134] [138] [139], and these properties may be the source of positive cognitive response. Their next generation of methylene blue derived aggregation inhibitor, TRx0237, is currently in phase III clinical trial. Tau aggregation inhibitors may demonstrate not only the ability to prevent PHF tangle mediated toxicity, but may also promote binding of tau back to microtubules. Currently, multiple drugs involved in preventing tau aggregation and promoting microtubule stability are in development [140]. 4.5. Directed Therapeutics: Microtubule Stabilization In vitro assays suggest that microtubule stabilizing agents, taxanes such as paclitaxel, have a protective effect on neurons subjected to beta-amyloid induced toxicity [94] [141]. The evaluation of microtubule stabilizing agents began in cancer research due to the implications of microtubules in cell pision, and thus, many known microtubule stabilizing agents, including paclitaxel, exhibit poor blood brain barrier permeability [142]. Investigations into blood brain barrier-permeable microtubule stabilizing agents have yielded several potential therapeutics. Epothilones show significant blood brain barrier permeability and exhibit microtubule stabilizing effects in mouse models such as normalization of microtubule density, restoration of axonal transport, and reduction in axonal dystrophy [143] [144]. As a result, the mice exhibited an increase in cognitive performance and a decrease in mortality rate. Multiple microtubule stabilizing agents have been evaluated in phase I clinical trials for AD in the past year. One such compound, BMS-241027, has demonstrated an ability to restore normal microtubule dynamics in multiple mouse model systems, with coinciding benefits on cognitive performance [145]. If the neurodegeneration encouraged by tau hyperphosphorylation is more heavily influenced by microtubule disintegration than tangle mediated toxicity, then microtubule-stabilizing agents may be a promising avenue for AD treatment. 4.6. Directed Therapeutics: Conclusion Tau directed therapies represent a quickly evolving ideology in the treatment of Alzheimer’s Disease. Anti-aggregation treatments for tau should generate a reduction in PHF tangle mediated toxicity, and microtubule stabilizing agents can restore cytoskeletal structure and axonal transport in neurons. A combinatorial approach J. Davis, R. Couch 116 might be sufficient to reverse the most direct effects of tauopathy. However, the changes in kinase/phosphatase activities in the AD brain present a pharmaceutical target that may have far reaching implications in other aspects of the disease beyond the hyperphosphorylation of tau. These proteins play integral roles in signal transduction and little is known as to how many of these functions are impaired in the AD system. 5. Other Alzheimer’s Defects and Related Therapeutics 5.1. Glucose Metabolism Defects in glucose metabolism have been illustrated as a characteristic of Alzheimer’s Disease. These faults are marked by reduced glucose utilization in early stages of AD and by downregulation of gene expression for proteins involved in oxidative phosphorylation [146]. The response is believed to be the result of damage to the mitochondria accrued either due to oxidative stress or as a protective response to downregulate energy metabolism in the nutrient-poor and oxygen-deficient AD brain [147]. One avenue of treating brain energy metabolism defects incorporates the use of antioxidants to prevent mitochondrial damage. At least 9 different anti-oxidants and multiple natural products with anti-oxidant effects have been evaluated in clinical trials for AD thus far. A second option being investigated is the use of alternative energy substrates for energy metabolism in the brain. AC-1204, a potential therapeutic currently in open clinical trials, induces ketosis for the purpose of providing a glucose alternative for brain energy metabolism [148]. A second treatment in phase I trial, GSK- 2981710, provides medium chain triglycerides as a potential energy source. Multiple insulin modulating agents have also been evaluated in clinical trials for AD. 5.2. Inflammation Recent research has pointed to inflammation, oxidative stress, and vascular degeneration as causative agents of the disease. The buildup of Aβ elicits an immune response from microglia and astrocytes. Microglia exhibit a limited phagocytotic ability for amyloid. However, activated astrocytes and microglia may stimulate the release of specific cytokines and chemokines to recruit peripheral macrophages for the purpose of Aβ clearance [149]. The response may stimulate the release of many cellular defense mechanisms such as the generation and localization of reactive oxygen species (ROS) and interleukins (IL-1β, IL-6, TNF-α and INF-γ) that are pro-inflammatory [149]. Aβ has also been shown to induce accumulation of major histocompatibility complex surface molecules and complement response [149]. Astrocyte activation is a known pathological response to AD, and astrocytes can phagocytose amyloid directly, but this action furthers the pro-inflammatory response [149]. Additionally, activation of astrocytes disrupts their normal function in neuronal support, resulting in local neuron depolarization and cytotoxic damage. This phenomenon can lead to neuronal degradation and disease progression if astrocyte activation continues to persist [149]. Previously, the decrease in AD rate amongst NSAID users [150] was mentioned in a discussion of NSAIDs as γ-secretase inhibitors. However, suppression of inflammation by NSAIDs may prevent the damaging inflammation described in this context. Multiple anti-inflammatory therapeutics, targeting cytokines, granulyte colony stimulating factor, tumor necrosis factor, and histamine related pathways are currently undergoing clinical trials as AD therapies. 5.3. Neurodegeneration Amyloid-β, complement, and oxidative damage have all been linked to the lysing and apoptosis of neurons in the brain. Examinations in synaptic density have revealed significant decreases in synaptic number per unit volume in both lamina III (-42%) and lamina V (-29%) of the human frontal cortex [151]. A negative correlation between synapse abundance and synapse size is also observed, likely as a compensatory response [151]. Significant decline in synaptic numbers in both the superior and middle temporal gyrus are also detected [152]. Lower hippocampal baseline volumes and higher atrophy rates are seen in patients with AD when compared to healthy controls, and whole brain atrophy rates are higher in patients with AD than in healthy controls or in persons afflicted with only mild cognitive impairment [153]. The selective loss of forebrain cholinergic neurons has been a known element of Alzheimer’s pathology since the 1970s [154]. The phenomenon correlates with the breakdown of cholinergic pathways in the cerebral cortex and basal forebrain of persons afflicted with AD [155]. Fittingly, 3 of the 4 FDA approved disease modifying therapies for Alzheimer’s (Donepezil, Rivastigmine, and Galantamine) are cholinesterase inhibitors that prevent the degradation of acetylcholine in the neural synapse J. Davis, R. Couch 117 [155] [156]. Treatment for neurodegeneration in Alzheimer’s Disease may involve the upregulation of neurotrophin levels in the brain. Neurotrophins such as brain derived neurotrophin factor (BDNF), neurotrophin-3 (NT3), and nerve growth factor (NGF) promote cell growth and cell survival upon binding to specific receptor tyrosine kinases on the neuron (TrkB, TrkC, and TrkA respectively) [157]. Binding of nerve growth factor to TrkA stimulates a signal transduction cascade that promotes neuron survival through inhibition of apoptotic proteins via the PI- 3K/AKT pathway [158]-[160], through MEK/MAPK pathway stimulation of anti-apoptotic (pro-survival) proteins [161]-[163], and through upregulation of CREB [162] [163]. The MEK/MAPK pathway has also been shown to protect sympathetic neurons from cytokine arabinoside induced apoptosis and cerebellar neurons from apoptosis due to oxidative stress [163]-[166]. Several studies have illustrated the protective effect of NGF in animal models. Mouse-NGF [167] and recombinant human NGF [168] have been shown to almost completely prevent reductions in the number and size of cholinergic neurons. Recombinant human NGF has also been shown to be active in the prevention of cholinergic neuron degradation in primates caused by age [169]. Current investigations are underway to identify methods of delivering neurotrophins to the brain via virus-mediated gene delivery [170] and upregulation of endogenous secretion [171]-[174]. 5.4. Novel Therapies Other avenues of treating Alzheimer’s Disease are being investigated in addition to the methods described above. Phosphodiesterase (PDE) inhibitors are currently being evaluated in clinical trials for AD. PDE inhibition would cause elevated levels of cAMP and cGMP in the brain, stimulating cAMP response element-binding protein (CREB). CREB promotes the gene expression of neurotrophins [175] and may play a role in long-term potentiation and long term memory formation [176]-[180]. Several mouse model studies of PDE inhibitors suggest potential efficacy for AD treatment [181]-[183]. Additionally, unique targets such as HMG-CoA reductase (implicated in cholesteremia), peroxisomes, angiotensin receptors, heperanase, and matrix metalloproteins have been investigated in clinical trials for AD. Hormone supplementation has been evaluated for effects on the disease and the potential of vitamin supplementation has also been explored. Thus, the approaches to treating Alzheimer’s appear to be broad and multifaceted. 6. Conclusion Much criticism surrounds the state of Alzheimer’s research as it relates to therapeutics, suggesting that the current approach to treating the disease focuses too narrowly on amyloid plaques and neurofibrillary tangles of hyperphosphorylated tau. However, current investigations attack nearly every known aspect of the etiology and symptoms that characterize AD. Greater success in treating the disease will require a greater understanding of disease progression and the elucidation of its biochemical origin. Many believe that amyloid- and tau-based treatments for Alzheimer’s Disease would provide better efficacy if administered before these morphological changes are able to produce significant cognitive impairment. New developments in brain imaging and earnest efforts to identify the earliest symptoms of Alzheimer’s-type cognitive decline will play a critical role in improving the field of Alzheimer’s treatment, providing greater chance of efficacy for existing therapies and new targets for rational drug design.
Strategizing the Development of Alzheimer’s Therapeutics Justin Davis, Robin Couch Department of Chemistry and Biochemistry, George Mason University, Manassas, VA, USA Email: [email protected] Received 10 June 2014; revised 25 July 2014; accepted 5 August 2014 Copyright © 2014 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Alzheimer’s Disease is a complex, progressive condition with symptoms that do not reveal themselves until significant changes to neuronal morphology have already occurred. The delayed manifestation of cognitive decline makes determination of the true etiological origins difficult. As a result, identification of ideal drug targets becomes seemingly impossible. The existing treatments for Alzheimer’s Disease may temporarily suppress the rate of cognitive decline, but do little to slow or halt neuronal decay. While many believe that the current approaches to identifying a cure for the disease are too narrow-minded, focusing heavily on the physical manifestations of the diseased brain such as amyloid plaques and neurofibrillary tangles, this review asserts the status of Alzheimer’s research as rational and multi-faceted. Keywords Drug Discovery, Alzheimer’s Disease, Target Identification, Therapeutics, Review 1. Introduction Alzheimer’s Disease (AD), the most common form of dementia, involves the progressive deterioration of cognitive and functional abilities, ultimately leading to the loss of bodily function and death. According to the World Health Organization, dementia affects 35.6 million people worldwide, with the figure expected to double by 2030 and triple by 2050 in the absence of a new therapy capable of preventing, halting, or slowing disease progression. Alzheimer’s Disease accounts for up to 70% of these cases, with an annual estimated worldwide cost of $600 US billion [1] [2]. In the United States, one in nine people over the age of 65 is believed to suffer from AD [3]. The earliest symptoms of AD include deficits in working memory and concentration. Visuospatial and visuoconstructional deficiencies as well as naming and language impairments also appear early in the course of the J. Davis, R. Couch 108 disease [4]. As AD progresses, changes in social and emotional behaviors occur, including agitation, psychosis, and depression. Later stages of AD are marked by intermediate and long-term memory loss, loss of self-sustainnability, and changes in sleep cycle. The final stages of AD entail the loss of control over bodily function and may often be characterized by a vegetative state [4]. Upon onset, life expectancy is reduced to a range of 4 to 12 years [4], correlating to a 67% reduction in expected life span for diagnoses at age 65 [5]. Alzheimer’s Disease is the sixth leading cause of death in the United States, causing over 80,000 deaths in 2010 [3]. The pathogenesis of Alzheimer’s Disease is still being elucidated and is often characterized by the development of beta-amyloid aggregates, tau hyperphosphorylation, changes in neuronal glucose metabolism, and neurodegeneration. The breakdown of neural connections eventually leads to dementia and loss of cognitive function. Therapeutics currently approved for the treatment of Alzheimer’s Disease help to temporarily mitigate symptoms and improve quality of life by affecting neurotransmission; however, these drugs are not disease-modifying and fail to address the etiological underpinnings of the disease. The following review will detail the current treatments for Alzheimer’s Disease and will present strategies for developing new, disease-modifying treatments based on various elements of known AD pathology. 2. Current Therapeutics: Pharmaceutical Treatment of Neurotransmitter Signaling Currently, the four FDA approved medications for AD treat the symptoms of the disease, mainly by acting on the neurotransmitter signaling. The hallmarking memory deficit of AD was linked to a substantial loss of cholinergic neurons in the basal forebrain and cerebral cortex as early as the 1980s [6] [7]. The noted deficit in cholinergic synapses led to the development of therapeutic agents designed to restore cholinergic neurotransmission via inhibition of acetylcholinesterase (AChE), an enzyme that hydrolyzes free acetylcholine in the synapse. While these agents have demonstrated the ability to slow the rate of cognitive decline, they have little effect on the pathogenesis of AD. Galantamine, a phenanthrene, is the weakest of the approved acetylcholinesterase inhibitors (IC50 400 - 800 nM [8] [9]) and is a competitive, reversible inhibitor of the enzyme [10]. The compound has also been shown to allosterically agonize the nicotinic acetylcholine receptor at low concentrations, with antagonistic effects at higher concentrations, likely through a competitive mechanism [11] [12]. This dual mode of enhancing acetylcholine signaling likely offsets its lower binding affinity for AChE, as galantamine produces similar positive effects on cognition, behavior, and general function when compared to the other approved acetylcholinesterase inhibitors [13] [14]. Donepezil, a piperidine, is a highly specific, non-competitive, reversible AChE inhibitor (IC50 6.7 nM [15]) that fits within the active site gorge of AChE without directly interacting with the catalytic triad of the enzyme [16]. The structural differences between donepezil and the other inhibitors contributes to its longer half-life (-73 hours versus 4 - 6 for rivastigmine and galantamine) [14]. In comparison with placebo, donepezil demonstrates a statistically significant improvement in cognitive function as well as positive behavioral modification [14]. Rivastigmine, a carbamate derivative, is a non-competitive, reversible AChE inhibitor (IC50 4.4 nM [15]) that inhibits both acetylcholinesterase and butyrylcholinesterase with similar affinity [17]. Rivastigmine remains bound to the enzyme for a greater amount of time than the other AChE inhibitors (10 hours versus minutes for donepezil) [18], as its carbamate moiety remains bound in the esteratic site of the enzyme following hydrolysis [18]. Rivastigmine generates improvements in cognitive function at low doses of 1 - 4 mg/day and highdoses of 6 - 12 mg/day. However, the higher doses required to produce enhancements in global assessments also correlate with a higher rate in discontinuation due to adverse effect [14]. Most comparative analyses investigating the effectiveness of the three acetylcholinesterase inhibitors suggest that all three compounds have similar efficacy in regards to cognitive function, behavior, and global outcome [14]. Meta-analysis performed by Hansen et al. in 2008 [19] proposes a greater behavioral response for patients treated with donepezil and a better global assessment with donepezil or rivastigmine, although the differences are minute. Overall, the three acetylcholinesterase inhibitors temporarily stabilize the rate of behavioral, cognitive, and functional decline. These effects last for a range of six months to one year, before cognition falls below baseline levels [20]. The remaining FDA approved treatment for Alzheimer’s disease, memantine, affects glutamate signaling through uncompetitive antagonism of NMDA (N-methyl-D-aspartate) receptors (IC50 0.3 - 10 μM, depending on J. Davis, R. Couch 109 membrane potential [21] [22]). The open channel blocker has a limiting effect on Ca2+ flux, with potential therapeutic effects of suppressing long term potentiation and limiting excitotoxicity [21]. Memantine also has a similar inhibitory effect on both α7 and α4β2 nicotinic acetylcholine receptors (IC50 - 10 μM [21] [22]), with opposite effects on cholinergic transmission than the acetylcholinesterase inhibitors approved as AD therapeutics. The compound shows efficacy in patients with moderate to severe AD when used as a monotherapy [23] [24] or in conjunction with the approved acetylcholinesterase inhibitors [24]. At least 24 compounds targeting acetylcholine signaling and 9 compounds targeting glutamate signaling (through NMDA or AMPA receptors) had been evaluated in clinical trials for Alzheimer’s as of May 2014. Additionally, at least 29 dopamine or serotonin modulators have been evaluated as well. In total, approximately one third of all clinical trials (phase I-IV) for the treatment of Alzheimer’s Disease target neurotransmitter signaling in the brain. These statistics suggest that while Alzheimer’s is often described by the etiological issues of amyloid plaques and tau hyperphosphorylation, the treatment of its symptomatic deficits in memory and cognitive decline is still a key component of attacking the disease and its symptoms. 3. Strategizing Therapeutics: The Amyloid Hypothesis 3.1. General Background Alzheimer’s Disease, as it was first described by Dr. Alois Alzheimer in 1907, is most often characterized by the accumulation of beta-amyloid plaques derived from the aggregation of small amyloid-β (Aβ) peptides in the brain. The amyloid-β peptide is generated by cleavage of membrane-bound amyloid precursor protein (APP), a protein involved in synaptogenesis [25]. The Aβ peptide is excised from APP holoprotein by β-APP cleaving enzyme (BACE or β-secretase) and γ-secretase [26] [27]. β-secretase acts extracellularly on APP, releasing a large fragment (sAPPβ) to the extracellular environment. The remaining 99 amino acid fragment (CTFβ) remains temporarily bound to the membrane [27]-[29]. The subsequent intracellular action of γ-secretase on CTFβ results in the generation of fragments Aβ40 and Aβ42, depending upon the site of cleavage [27]-[30] (Figure 1). The conformation of the longer fragment, Aβ42, differs from that of Aβ40 [31]-[33], causing it to be the “stickier” of the two aggregation-prone beta-amyloid peptides. As a result, Aβ42 is more often implicated in the formation of the amyloid plaques [30]. In healthy inpiduals, γ-secretase generates Aβ40 8 to 9 times more often than Aβ42 [27]-[34]. Additionally, neither β-amyloid is the most prevalent APP cleavage product found in the brain due to APP processing by α-secretase [27] [34] [35]. Although β-secretase activity and its downstream products are significant due to their roles in amyloid plaque formation, β-secretase acts on only 10% of the total APP in healthy cells [27] [35]. Most often, APP is cleaved by α-secretase, generating sAPPα extracellularly and an 83 amino acid fragment, CTFα [26] [27] [35]. Intracellular cleavage of CTFα by γ-secretase generates a p3 fragment that is not prone to plaque formation in the fashion of Aβ [27]. However, while the amount of Aβ42 present in healthy brains is minimal, AD is marked by a suppression of α-secretase activity and a gain in β-secretase activity that enhances plaque formation through increased production of amyloid-β [35]. The driver for this change in relative secretase activities is unknown. Figure 1. Processing of amyloid precursor protein (APP). Cleavage by β-secretase and subsequent cleavage by γ- secretase generates amyloid-β peptides, 40 to 42 amino acids in length. Aβ42 is more prone to aggregation and plaque formation. J. Davis, R. Couch 110 Aggregation of Aβ occurs in two phases, with soluble multimers preceding the development of fibrillar deposits [36] [37]. The two multimeric forms trigger different toxic responses and are recognized by different antibodies [27]. The exact mechanism of action for Aβ induction of neurodegeneration is unknown, but smaller, soluble Aβ aggregates are believed to be the form most relevant to amyloid related toxicity in AD [27] [37]. Additionally, Aβ aggregates can deposit in the walls of blood vessels, causing intracerebral hemorrhaging [38]. Most believe that the development of beta-amyloid aggregates is the initiating step in the disease, and thus, tau hyperphosphorylation, excitotoxicity, changes in glucose metabolism, and neurodegeneration may all be downstream results of amyloid-β accumulation. Prevention of amyloid-β accumulation requires the degradation or clearance of the peptide. The enzymes neprilysin (NEP) and insulin degrading enzyme (IDE) have been implicated as the proteins responsible for degrading Aβ in the cell [27] [39] [40]. Neither protease is specific for Aβ, and IDE shows 20-fold greater binding affinity for insulin than for Aβ [27] (thus, insulin may act as a competitive inhibitor of IDE degradation of Aβ, serving as a potential link between insulin-related diseases and AD). Much Aβ remains undegraded, and some is transported through the blood brain barrier by low-density lipoprotein receptor-related protein (LRP) on the brain side and the receptor for advanced glycation end products (RAGE) on the blood side [27] [41]. Aβ that remains unaffected by degradation and transport has greater chance to form aggregates. The perceived significance of amyloid-β to AD was heightened by the discovery of genetic mutations and risk alleles that relate directly to amyloid-β and its clearance from the cell. Autosomal dominant mutations implicated in familial, early onset AD, have been detected in genes for presenilin 1 and 2 (PSEN 1 and PSEN 2) [26] [42] [43], which code for the catalytic unit of the γ-secretase complex. The mutations of PSEN 1 and PSEN 2 alter the cleavage activities of γ-secretase, resulting in a higher ratio of Aβ42 to Aβ42 [26] [42]. Autosomal dominant mutations in the gene for amyloid precursor protein that cause changes in processing have also been implicated in promoting the generation of Aβ42 [44] [45]. Additionally, three different apolipoprotein E (APOE) alleles are common in the population. APOE2, APOE3, and APOE4 have been correlated to AD with APOE2 delaying the onset of disease, APOE3 being neutral, and APOE4 decreasing the age of onset [26]. The mechanism of action is believed to involve direct removal of amyloid-β [46], as APOE2, APOE3, and APOE4 have decreasing effectiveness in Aβ clearance [26] [47]. As the only known genetic mutations and risk alleles for AD relate directly to amyloid-β, and not to other known features of AD, inhibiting Aβ production and preventing its aggregation is a rational approach to treating the disease. 3.2. Directed Therapeutics: Gamma-Secretase Inhibition and Modulation Gamma secretase has been evaluated as a target to minimize beta-amyloid production with minimal success. The aspartylintramembranal protease is composed of Aph1, nicastrin, Pen2, and presenilin, the gene product implicated in two of the three major genes associated with familial AD [48]. Presenilin serves as the catalytic component of γ-secretase. However, the protease also acts on other transmembrane proteins, including p75 neurotrophin receptor, ErbB4, N-cadherin, and Notch [49] [50]. Proteolytic cleavage of Notch by presenilin promotes its nuclear translocation, initiation of signal transduction cascades, and transcription regulation, inhibiting downstream modulation of cell differentiation and proliferation [51] [52]. As expected, γ-secretase inhibitors (GSIs), such as MK-0752, have been evaluated in clinical trials as anti-cancer agents due to its depressive effects on Notch [53]. Complete inhibition of γ-secretase functioning, through binding to presenilin fragments involved in protease activity, results in dramatic side effects. Semegestat, the poster-drug for detractors of the amyloid hypothesis, advanced to phase III clinical trials as a γ-secretase inhibitor, but led to significant worsening of cognitive function despite reducing beta-amyloid load [50]. The decline in cognitive function may be linked to observed changes in hippocampal signaling [54]. The mechanism is unknown, but may be attributed to downstream effects on presenilin’s other protease targets, including Notch, which has been implicated in the regulation of dendritic development [55] and axon arborization (branching) [56]. Noted hematologic toxicity, skin toxicity, and gastrointestinal toxicity, likely induced by changes in intestinal epithelium due to Notch inhibition [57], brought a premature end to the clinical trial, and a similar fate was shared by avagacestat, another γ-secretase inhibitor. Avagacestat was also linked to an increase in nonmelanoma skin cancer during phase II clinical trials [58], causing further debate as to the viability of γ-secretase as a suitable drug target for AD. These results have J. Davis, R. Couch 111 brought about a change in the design of γ-secretase inhibitors, from compounds aimed at total complex inhibition, to compounds designed to modulate γ-secretase. These γ-secretase modulators (GSMs) inhibit γ-secretase through allosteric mechanisms that permit the other actions of presenilin. The overarching aim of GSMs is to alter the γ-secretase complex in a way that favors the production of shorter forms of beta-amyloid that are less prone to aggregation, such as Aβ38,without interfering with γ-secretase action on other targets such as Notch. In one potential mechanism, noncompetitive binding to a novel site on γ-secretase induces a conformational change that shifts the cleavage site [59]. Alternatively, the compound may act directly with APP, altering the availability of the Aβ42 and Aβ40 cleavage sites [48] [60]. Over the past decade, several nonsteroidal anti-inflammatory drugs (NSAIDs), including ibuprofen, have been evaluated as a treatment for AD due to an observed reduction in AD prevalence amongst NSAID users. These compounds, serving as the first generation of GSMs, demonstrated effects of γ-secretase modulation in vitro, with a decrease in Aβ42 correlating to an increase in Aβ38. Tarenflurbil, R-fluriprofen, failed phase III clinical trials in 2009, showing no effect on cognitive decline [61]. Although the compound showed an effect on Aβ42 in cell based assays, similar effects were not observed in patients despite dose dependent blood-brain barrier penetration [62]. More recent methods of designing GSMs have involved modifications to existing NSAID structure as well as designing new compounds altogether, with the ultimate goal of reducing Aβ42 levels in the human brain. In general, γ-secretase modulators have reduced potency compared to γ-secretase inhibitors in terms of overall reduction of Aβ42 [63]. As such, the design of blood brain barrier-permeable molecules that exhibit minimal toxicity at concentrations sufficient for preventing Aβ42 production is the greatest barrier in the γ-secretase directed therapies. 3.3. Directed Therapeutics: Beta-Secretase Inhibitors Another avenue of reducing amyloid load in the brain is by limiting Aβ production using β-secretase (BACE-1) inhibitors. These compounds reduce the amount of Aβ detected in plasma and cerebrospinal fluid (CSF) after administration [50] [64] [65] and have generated cognitive improvements in mouse models [66]. The side effects of BACE-1 inhibition have been debated, with abnormalities in BACE-1 negative mice ranging from minimal [64] [67] to the development of memory deficits, seizures, hypomyelination, and changes in axonal guidance [68]-[70]. BACE-1 inhibitors have become prevalent in clinical trials since the first phase I study of CTS21166 in 2008. This compound caused a dose-dependent depletion of plasma Aβ by up to 80%, with full recovery occurring 144 hours after administration [64]. A second BACE-1 inhibitor, LY2886721, demonstrated reductions in Aβ40 and Aβ42 in a phase I study; however, phase II trials were terminated prematurely due to toxicity in the liver. A relationship between the BACE-1 inhibition mechanism and liver-related side effects is yet to be identified [65]. MK-8931, the first BACE-1 inhibitor to enter a phase III trial, should demonstrate the effects of BACE-1 inhibitors on cognitive function. The compound has, like the other clinically tested BACE-1 inhibitors, led to reductions in Aβ following administration [50]. MK-8931 is being evaluated in cohorts with mild and prodromal AD, and the results should shed light on the potential advantages of targeting Aβ at early disease stages. In the past five years, at least four BACE-1 inhibitors have completed clinical trials as Alzheimer’s therapeutics and two are currently in testing. 3.4. Directed Therapeutics: Active Immunization Active immunization against amyloid-β has been attempted since the late 1990s with limited success. Theoretically, the strategy would allow a patient’s immune response to produce oligoclonal antibodies for beta-amyloid that are persistent. The first major attempt, AN1792, utilized the complete Aβ42 peptide in combination with the QS-21 adjuvant to elicit an immune response capable of clearing amyloid-β. In the case of active and passive immunization, Aβ clearance appears to depend on a microglial FcR dependent mechanism that stimulates phagocytosis [71]. Mouse models showed not only a reduction in amyloid load, but also a suppressive effect on cognitive decline, corresponding to a reduction in neuritic dystrophy and angliosis [72]. However, Phase II clinical trials in 2001 were unsuccessful. Although the treatment successfully caused a decrease in total amyloid-β load (analyzed post-mortem), the active immunization method yielded no effect on cognitive performance or survival over the 6 year follow-up period. Additionally, meningoencephalitis occurred in 18 of the 80 patients studied, causing the trial to be ceased prematurely [50] [73]. More recent approaches to active vaccination have utilized only the N-terminal fragment of amyloid-β and have varied the use and type of adjuvants in order to promote amyloid clearance without inducing excessive in- J. Davis, R. Couch 112 flammation. UB 311 employs two synthetic peptides linked to the N-terminal 14 amino acid fragment of amyloid-β and is provided with a mineral salt adjuvant. Mouse studies showed diminished amyloid-β load in the cortex and hippocampus and corresponding improvements in learning and short term memory. Initial results of a clinical trial involving 19 patients revealed elevated antibody titers over a 48 week period, as well as measurable improvement in cognitive function, with no reported brain inflammation noted thus far [74]. A second vaccine, AC-24, utilizes an N-terminal 15 amino acid fragment in a liposomal surface. AFFiris has employed an approach of using short peptide fragments of 8 amino acids or less in order to minimize T cell response. CAD106 also incorporates this tactic and successfully causes an increase in antibody titers without eliciting T cell response [75]. The vaccination approach for reducing amyloid load is becoming more prevalent, with the focus resting on maximizing antibody generation and amyloid clearance whilst minimizing inflammation. Adjuvant selection, fragment choice, and the stage of AD in which vaccination is delivered will all play roles in the success of this treatment method. 3.5. Directed Therapeutics: Passive Immunization Passive immunization using humanized IgG antibodies against beta-amyloid provides a second option to promote Aβ clearance without eliciting T-cell mediated inflammation. The most glaring challenge in a passive antibody mediated therapy would likely by delivering the antibody to the brain across the blood brain barrier. As early as 2000, passive immunization was displaying efficacy in mouse models by reducing amyloid load, limiting neuritic dystrophy, and producing a marked improvement in cognitive function [76]-[78]. Successes in the mice model go against expectations for blood brain barrier permeability. The first attempts at passive immunization resulted in elevated levels of plasma Aβ and little Aβ bound to antibody in the brain. The phenomenon lead to the development of the “peripheral sink” hypothesis. In this mechanism, equilibrium draws the amyloid-β peptide into the plasma after plasma-Aβ levels are reduced by the antibody [76]. This result was observed not only in the mouse model, but also in the case of bapineuzumab, the first monoclonal anti-Aβ antibody to reach Phase II clinical trials. Bapineuzumab, a humanized monoclonal anti-Aβ antibody directed at the N-terminus of Aβ, exhibits ApoE dependent reduction in beta-amyloid, with carriers of ApoE4 showing minimal response [50] [76]. Several cases of mild vasogenic edema were observed, more commonly in ApoE4 carriers, but these cases were not severe enough to cause trial cessation. The correlation between the peripheral sink hypothesis and vasogenic edema do suggest the possibility of an inflammation-based mechanism of action that depends upon enhanced blood brain barrier permeability. In terms of cognition, a minimal clinical effect was observed, with a 5% - 6% improvement compared to placebo at the end of 78 weeks (ADAS-Cog and DAD scores respectively) [76]. Phase III clinical trials of bapineuzumab have been discontinued due to failure to show significant changes in cognitive end points, but the compound did elicit a decrease in amyloid burden and CSF phosphorylated tau [79]. The other passive immunotherapy to have completed phase III clinical trials, Solanezumab, is directed at a central epitope of Aβ, favoring the removal of free Aβ or small soluble fibrils over plaques. This compound produced no effect on phosphorylated tau and failed phase III due to inability to meet cognitive end points. More recent attempts at passive immunization are derived from monoclonal antibodies developed against protofibrillar species (BAN2401, Crenezumab, SAR228810) [79]. Clinical outcome data from trials involving these treatments should be released in the upcoming years, providing more evidence as to the potential efficacy of passive immunization. 3.6. Directed Therapeutics for Beta-Amyloid: Conclusion Thus far, beta-amyloid directed therapies have failed to slow or halt the progression of Alzheimer’s Disease. However, the recent progress in immunization and secretase-inhibition do provide hope for a potential beta-amyloid targeted cure. Development in active immunization, passive immunization, γ-secretase inhibition, and β-secretase inhibition is currently ongoing, providing a multifaceted approach for preventing beta-amyloid accumulation. 4. Strategizing Therapeutics: Tau Hypothesis 4.1. General Background Failures of amyloid-based treatments to have a significant impact on the progression of Alzheimer’s Disease J. Davis, R. Couch 113 have called into question the role of amyloid plaque formation as a causative agent of AD. Clinically, loss of brain mass and the progression of dementia correlate only weakly to the quantity and distribution of amyloid-β in the brain, and amyloid load may be present in up to 30% of the elder population with no signs of the Alzheimer’s phenotype [27] [80] [81]. Instead of being the driving force behind the AD neurodegeneration, many have postulated that the formation of amyloid plaques is just one of many secondary events that occur in the progression of the disease. The Tau hypothesis seeks to provide an alternative rationale, attributing Alzheimer’s Disease to the formation of neurofibrillary tangles composed of hyperphosphorylated tau [26] [27] [30]. The causes of excessive tau phosphorylation are still unverified, but toxicity induced by Aβ, oxidative stress, and inflammation may induce upregulation of tau kinases, downregulation of phosphatases, and modification to tau directly (post-translational modifications of tau are discussed in the next section) [82] [83]. Tau is a soluble microtubule-associated protein and has stabilizing effects on microtubulin assembly [84]-[86]. Hyperphosphorylation of tau, an indicator of AD, inhibits the association of tau with the microtubules and induces the formation of tau aggregates [87]-[89]. The microtubule network plays a critical role in neuronal morphology and the transport of signaling molecules, trophic factors, and organelles [85] [86]. Tau mutations and hyperphosphorylation have been correlated to a reduction in microtubule density and fast axonal transport in mouse models [83]. In a recent study, providing a microtubule-stabilizing drug to tau-transgenic mice with microtubule and motor deficits led to improvement in microtubule density and fast axonal transport as well as an improvement in motor function [83]. The gene for tau (tubulin-associated unit) is composed of 16 exons. The protein is generally described as four segments (Figure 2): an acidic N-terminal segment produced from exons 1 - 5, a proline rich region from exon 7 and part of exon 9, the microtubule association region composed of exons 9 - 12, and a C-terminal portion from exon 13. Products from exon 6, 8 and 14 are not observed. Exons 2, 3, and 10 undergo alternative splicing, resulting in six observed isoforms with varying affinity for microtubulin. These isoforms are designated 4R/2N, 4R/1N, 3R/2N, 4R/0N, 3R/1N, and 3R/0N. R describes the exons in the repeat domain region, with 4R designating the presence of exon 10 and 3R designating absence. N describes the Nterminal region, with 2N containing both exon 2 and 3, 1N containing only exon 2, and 0N containing neither exon 2 nor exon 3 [82] [90]-[92]. The repeat domains, responsible for microtubule binding, contain the conserved sequence motif, KXGS. The serine residue in this motif is prone to phosphorylation, resulting in destabilization of the tau protein. Tau expression appears to be dynamic, with 3R tau being more prevalent during development, allowing for enhanced plasticity corresponding to decreased cytoskeleton support. Adult neurons coincide with a closer to 1:1 ratio of 4R:3R and decreased hyperphosphorylation, providing more tau-assisted cytoskeletal stabilization. In disease, a change in 4R:3R ratio is observed [92] [93]. The effect of tau hyperphosphorylation on the microtubule network is one of only two potential causes of tau induced neurodegeneration. Neurofibrillary tangles occur when hyperphosphorylated tau forms aggregates with tau, microtubule associated proteins 1 and 2, and ubiquitin [82] [88]. Straight or paired helical forms of these aggregates exist with a notable toxic effect on the cellular environment [82]. The abnormally phosphorylated tau, PHFtau, can aggregate to form paired helical filaments [94] [95]. However, only 10% of PHFtau is found in neurofibrillary tangles, with the remaining portion deposited in neuronal processes, appearing as dystrophic neuritis with swollen axons and dendrites [94]. Recent animal model studies suggest that microtubule stability is sufficient for cognitive improvement, regardless of the continued presence of neurofibrillary tangles [94]. This evidence points to microtubule dissociation as the key component of disease progression resulting from hyperphosphorlyated tau. 4.2. The Role of Kinases and Phosphatases in AD Pathology Three classes of tau kinases phosphorylate the tau protein, including tyrosine-specific kinases, proline directed kinases (PDKs), and non-proline directed kinases. The major proline directed kinases acting on tau include glycogen synthase kinase (GSK-3β), cyclin dependent kinase (CDK5), and mitogen-activated protein kinases (MAPKs). Relevant non-PDK kinases are tau-tubulin kinase, casein kinase, dual-specificity tyrosine phosphorylation and regulated kinase (DYRK1A), microtubule affinity-regulating kinases, PKAA, PKB/Akt, PKC, PKN, and Calmodulin-dependent protein kinase (CaMKII) [92]. Hyperphosphorylated tau contains up to 10 phosphates per molecule, representing a 3 to 4 fold increase compared to tau in healthy inpiduals. Of the 85 serine, J. Davis, R. Couch 114 Figure 2. The largest tau gene product, 4R/2N, is indicated by the presence of exon 10 and exons 2 and 3. Six isoforms of the gene exist. The 4R/1N isoform is marked by the absence of exon 3 and 4R/0N contains neither exon 3 nor exon 2. The 3R isoforms have lower affinity for microtubulin and are marked by the absence of exon 10. threonine, and tyrosine residues that could serve as targets for phosphorylation by esterification, as many as 45 are exclusive targets for hyperphosphorylation in AD [96]-[100]. GSK-3β, CDK5, CK1, and PKA demonstrate the ability to generate all but eleven of these phosphorylation events, with much overlap in coverage amongst the kinases [96] [99] [101]-[103]. In AD, changes in expression and activity of GSK-3β, CDK5, DYRK1A, p38, and CK1 have been reported [92]. The sequence of phosphorylation events could provide insight into potential therapies. Phosphorylation by DYRK1A and CDK5 precede subsequent GSK-3β phosphorylation in vitro. Additionally, CK1 and c-Abl activate CDK5 phosphorylation events [93]. PKB, PKC, and PKN appear to inhibit phosphorylation by GSK-3β [104]. GSK-3β receives enhanced attention because of its implication in long term potentiation [100] [105] [106]. The kinase plays a role in NMDA receptor trafficking and therefore glutamate signaling, implying a potential mechanism for excitotoxicity [100] [107]. Additionally, a region of presenilin 1 that interacts with GSK-3β exhibits binding affinity for the microtubule binding domains of tau, providing a mechanism in which presenilin could facilitate an interaction between tau and the kinase and suggesting another means for presenilin mutations to induce neurodegeneration [90]. GSK-3β may also play an integral role in amyloid-induced tauopathy. In mouse models, amyloid peptides induce tyrosine autophosphorylation of GSK-3β at Y216, activating the kinase [100] [108]. In the frontal cortex of the AD brain, pY216-GSK3 has been observed as co-localized with tau protein, supporting this model of GSK activation leading to tau hyperphosphorylation [109] [110]. Although some debate surrounds whether or not the development of beta-amyloid deposits absolutely occurs before tauopathy in every case of AD [111] [112], models suggest that tau reduction prevents amyloid-induced excitotoxicty [113] [114] and defects in axonal transport [115]. Phosphatases, specifically protein phosphatase-2A (PP2A), play a critical role in tau hyperphosphorylation. The region of tau responsible for binding microtubules shows high affinity for PP2A, alluding to a mechanism in which unbound tau serves as a substrate for PP2A dephosphorylation, promoting subsequent binding to microtubules [90]. The Alzheimer’s brain exhibits a 50% reduction in PP2A activity and a 20% increase in PP2A inhibitors PP2A 1I and PP2A 2I [92] [116]. Expression of mRNA coding for the regulatory subunits and catalytic subunit of PP2A is downregulated in the hippocampus [117]. In model systems, the addition of PP2A reduces tau aggregates and encourages tau binding to microtubules [92] [118]. Methylation of the catalytic subunit of PP2A is required for activity, yet the responsible methyltransferase (LCMT-1) is under expressed in AD brains [119] [120]. Additionally, PP2A phosphatase activator, an enzyme that activates PP2A by removing an inhibitory phosphate, is also downregulated in AD [120] [121]. Glycosylation of tau, particularly O-glycosylation by N-acetyl-glucosamine, is another posttranslational modification that may impact tau aggregation and association with microtubules. Conflicting descriptions of the glycosylation state of tau and its ramifications exist in the literature. Multiple studies have described glycosylation of tau in PHF tangles with no glycan observed in normal tau [122] [123]. These reports also suggest that glycosylation favors association into filaments, and that deglycosylation is sufficient for conversion into linear bundles, with dephosphorylation and deglycosylation having a restorative effect on tau binding to microtubulin [122] [123]. Data also indicates that glycosylation of tau in the AD brain precedes tau hyperphosphorylation [124] [125]. Such a result might imply that glycosylation makes tau a better substrate for kinases or a poorer substrate for phosphatases, and in vitro assays do support this hypothesis [125]. However, recent evidence suggests that O-linked N-acetylglucosamine is more prevalent in healthy controls than in AD brains, and that glycosylation has an inhibitory effect on phosphorylation [126] [127]. In 2012, Yuzwa et al. performed a mouse model study [128] using an inhibitor of O-GlcNAcase that resulted in increased levels of O-glycosylated tau, decreases in tau aggregation, and decreases in neuronal cell loss. Interestingly, no changes in tau phosphoryla- J. Davis, R. Couch 115 tion were observed. The results conflict with evidence suggesting that glycosylation promotes aggregation; however, the authors’ in vitro analysis supports their claim of O-GlcNAc inhibiting oligomerization of tau. While many advocate the possibility of O-GlcNAcase inhibitors as a potential therapeutic for the treatment of AD, questions remain as to the consistency of tau glycosylation effects in AD etiology. 4.3. Directed Therapeutics: Kinase Inhibition The use of GSK-3β inhibitors as a means of treating AD is being evaluated. In mouse models, inhibition of GSK-3β resulted in reduced tau phosphorylation, tauopathy, neurodegeneration, neurofibrillary tangle formation, and neuronal loss [93]. Two specific GSK-3β inhibitors, NP031112 and tideglusib, have completed phase II trials with results yet to be published. In a pilot study, tideglusib appeared to elicit a positive cognitive response with minimal toxicity, although sample size was inefficient to determine true efficacy [129]. Cerebrolysin, a compound with regulatory effects on GSK-3β and CDK5 has also been involved in clinical trials as a combinatorial therapy with the approved acetylcholinesterase, donepezil. In a small sample size, cerebrolysin showed similar improvements as donepezil in cognitive performance, with a combinatorial approach showing enhanced gain over either inpidual treatment [130]. A c-Src/Abl kinase inhibitor, saracatinib, is currently in phase I trial, and more work targeting tau-related kinases is ongoing. 4.4. Directed Therapeutics: Tau Aggregation Inhibition An alternative method of treating tauopathy is through the use of tau aggregation inhibitors. In vitro assays and mouse model studies propose methylthionium chloride (methylene blue) as a compound capable of reducing aggregation of phosphorylated tau [131]-[133]. TauRx has developed multiple aggregation inhibitors that are derivatives of methylthionium chloride. The first of these compounds, TRx0014 has completed phase II clinical trials. Although the results pertaining to tau aggregation have yet to be published, preliminary data suggests a statistically significant delay in the rate of cognitive decline [134]. However, methylene blue has been indicated as an effector of butyrylcholinesterase activity [135], monoamine signaling [136] [137] and glutamate signaling [131] [134] [138] [139], and these properties may be the source of positive cognitive response. Their next generation of methylene blue derived aggregation inhibitor, TRx0237, is currently in phase III clinical trial. Tau aggregation inhibitors may demonstrate not only the ability to prevent PHF tangle mediated toxicity, but may also promote binding of tau back to microtubules. Currently, multiple drugs involved in preventing tau aggregation and promoting microtubule stability are in development [140]. 4.5. Directed Therapeutics: Microtubule Stabilization In vitro assays suggest that microtubule stabilizing agents, taxanes such as paclitaxel, have a protective effect on neurons subjected to beta-amyloid induced toxicity [94] [141]. The evaluation of microtubule stabilizing agents began in cancer research due to the implications of microtubules in cell pision, and thus, many known microtubule stabilizing agents, including paclitaxel, exhibit poor blood brain barrier permeability [142]. Investigations into blood brain barrier-permeable microtubule stabilizing agents have yielded several potential therapeutics. Epothilones show significant blood brain barrier permeability and exhibit microtubule stabilizing effects in mouse models such as normalization of microtubule density, restoration of axonal transport, and reduction in axonal dystrophy [143] [144]. As a result, the mice exhibited an increase in cognitive performance and a decrease in mortality rate. Multiple microtubule stabilizing agents have been evaluated in phase I clinical trials for AD in the past year. One such compound, BMS-241027, has demonstrated an ability to restore normal microtubule dynamics in multiple mouse model systems, with coinciding benefits on cognitive performance [145]. If the neurodegeneration encouraged by tau hyperphosphorylation is more heavily influenced by microtubule disintegration than tangle mediated toxicity, then microtubule-stabilizing agents may be a promising avenue for AD treatment. 4.6. Directed Therapeutics: Conclusion Tau directed therapies represent a quickly evolving ideology in the treatment of Alzheimer’s Disease. Anti-aggregation treatments for tau should generate a reduction in PHF tangle mediated toxicity, and microtubule stabilizing agents can restore cytoskeletal structure and axonal transport in neurons. A combinatorial approach J. Davis, R. Couch 116 might be sufficient to reverse the most direct effects of tauopathy. However, the changes in kinase/phosphatase activities in the AD brain present a pharmaceutical target that may have far reaching implications in other aspects of the disease beyond the hyperphosphorylation of tau. These proteins play integral roles in signal transduction and little is known as to how many of these functions are impaired in the AD system. 5. Other Alzheimer’s Defects and Related Therapeutics 5.1. Glucose Metabolism Defects in glucose metabolism have been illustrated as a characteristic of Alzheimer’s Disease. These faults are marked by reduced glucose utilization in early stages of AD and by downregulation of gene expression for proteins involved in oxidative phosphorylation [146]. The response is believed to be the result of damage to the mitochondria accrued either due to oxidative stress or as a protective response to downregulate energy metabolism in the nutrient-poor and oxygen-deficient AD brain [147]. One avenue of treating brain energy metabolism defects incorporates the use of antioxidants to prevent mitochondrial damage. At least 9 different anti-oxidants and multiple natural products with anti-oxidant effects have been evaluated in clinical trials for AD thus far. A second option being investigated is the use of alternative energy substrates for energy metabolism in the brain. AC-1204, a potential therapeutic currently in open clinical trials, induces ketosis for the purpose of providing a glucose alternative for brain energy metabolism [148]. A second treatment in phase I trial, GSK- 2981710, provides medium chain triglycerides as a potential energy source. Multiple insulin modulating agents have also been evaluated in clinical trials for AD. 5.2. Inflammation Recent research has pointed to inflammation, oxidative stress, and vascular degeneration as causative agents of the disease. The buildup of Aβ elicits an immune response from microglia and astrocytes. Microglia exhibit a limited phagocytotic ability for amyloid. However, activated astrocytes and microglia may stimulate the release of specific cytokines and chemokines to recruit peripheral macrophages for the purpose of Aβ clearance [149]. The response may stimulate the release of many cellular defense mechanisms such as the generation and localization of reactive oxygen species (ROS) and interleukins (IL-1β, IL-6, TNF-α and INF-γ) that are pro-inflammatory [149]. Aβ has also been shown to induce accumulation of major histocompatibility complex surface molecules and complement response [149]. Astrocyte activation is a known pathological response to AD, and astrocytes can phagocytose amyloid directly, but this action furthers the pro-inflammatory response [149]. Additionally, activation of astrocytes disrupts their normal function in neuronal support, resulting in local neuron depolarization and cytotoxic damage. This phenomenon can lead to neuronal degradation and disease progression if astrocyte activation continues to persist [149]. Previously, the decrease in AD rate amongst NSAID users [150] was mentioned in a discussion of NSAIDs as γ-secretase inhibitors. However, suppression of inflammation by NSAIDs may prevent the damaging inflammation described in this context. Multiple anti-inflammatory therapeutics, targeting cytokines, granulyte colony stimulating factor, tumor necrosis factor, and histamine related pathways are currently undergoing clinical trials as AD therapies. 5.3. Neurodegeneration Amyloid-β, complement, and oxidative damage have all been linked to the lysing and apoptosis of neurons in the brain. Examinations in synaptic density have revealed significant decreases in synaptic number per unit volume in both lamina III (-42%) and lamina V (-29%) of the human frontal cortex [151]. A negative correlation between synapse abundance and synapse size is also observed, likely as a compensatory response [151]. Significant decline in synaptic numbers in both the superior and middle temporal gyrus are also detected [152]. Lower hippocampal baseline volumes and higher atrophy rates are seen in patients with AD when compared to healthy controls, and whole brain atrophy rates are higher in patients with AD than in healthy controls or in persons afflicted with only mild cognitive impairment [153]. The selective loss of forebrain cholinergic neurons has been a known element of Alzheimer’s pathology since the 1970s [154]. The phenomenon correlates with the breakdown of cholinergic pathways in the cerebral cortex and basal forebrain of persons afflicted with AD [155]. Fittingly, 3 of the 4 FDA approved disease modifying therapies for Alzheimer’s (Donepezil, Rivastigmine, and Galantamine) are cholinesterase inhibitors that prevent the degradation of acetylcholine in the neural synapse J. Davis, R. Couch 117 [155] [156]. Treatment for neurodegeneration in Alzheimer’s Disease may involve the upregulation of neurotrophin levels in the brain. Neurotrophins such as brain derived neurotrophin factor (BDNF), neurotrophin-3 (NT3), and nerve growth factor (NGF) promote cell growth and cell survival upon binding to specific receptor tyrosine kinases on the neuron (TrkB, TrkC, and TrkA respectively) [157]. Binding of nerve growth factor to TrkA stimulates a signal transduction cascade that promotes neuron survival through inhibition of apoptotic proteins via the PI- 3K/AKT pathway [158]-[160], through MEK/MAPK pathway stimulation of anti-apoptotic (pro-survival) proteins [161]-[163], and through upregulation of CREB [162] [163]. The MEK/MAPK pathway has also been shown to protect sympathetic neurons from cytokine arabinoside induced apoptosis and cerebellar neurons from apoptosis due to oxidative stress [163]-[166]. Several studies have illustrated the protective effect of NGF in animal models. Mouse-NGF [167] and recombinant human NGF [168] have been shown to almost completely prevent reductions in the number and size of cholinergic neurons. Recombinant human NGF has also been shown to be active in the prevention of cholinergic neuron degradation in primates caused by age [169]. Current investigations are underway to identify methods of delivering neurotrophins to the brain via virus-mediated gene delivery [170] and upregulation of endogenous secretion [171]-[174]. 5.4. Novel Therapies Other avenues of treating Alzheimer’s Disease are being investigated in addition to the methods described above. Phosphodiesterase (PDE) inhibitors are currently being evaluated in clinical trials for AD. PDE inhibition would cause elevated levels of cAMP and cGMP in the brain, stimulating cAMP response element-binding protein (CREB). CREB promotes the gene expression of neurotrophins [175] and may play a role in long-term potentiation and long term memory formation [176]-[180]. Several mouse model studies of PDE inhibitors suggest potential efficacy for AD treatment [181]-[183]. Additionally, unique targets such as HMG-CoA reductase (implicated in cholesteremia), peroxisomes, angiotensin receptors, heperanase, and matrix metalloproteins have been investigated in clinical trials for AD. Hormone supplementation has been evaluated for effects on the disease and the potential of vitamin supplementation has also been explored. Thus, the approaches to treating Alzheimer’s appear to be broad and multifaceted. 6. Conclusion Much criticism surrounds the state of Alzheimer’s research as it relates to therapeutics, suggesting that the current approach to treating the disease focuses too narrowly on amyloid plaques and neurofibrillary tangles of hyperphosphorylated tau. However, current investigations attack nearly every known aspect of the etiology and symptoms that characterize AD. Greater success in treating the disease will require a greater understanding of disease progression and the elucidation of its biochemical origin. Many believe that amyloid- and tau-based treatments for Alzheimer’s Disease would provide better efficacy if administered before these morphological changes are able to produce significant cognitive impairment. New developments in brain imaging and earnest efforts to identify the earliest symptoms of Alzheimer’s-type cognitive decline will play a critical role in improving the field of Alzheimer’s treatment, providing greater chance of efficacy for existing therapies and new targets for rational drug design.
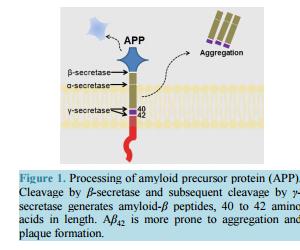 1
1
